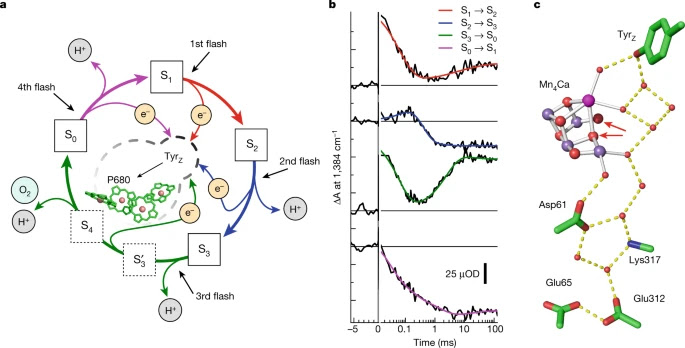
Reaction cycle of photosynthetic oxygen evolution. figure 1 a, Model of the S-state cycle with sequential electron and proton removal from the oxygen-evolving site10,11,50. Starting in the dark-stable S1 state, each laser flash initiates oxidation of the primary chlorophyll donor (P680+ formation) followed by electron transfer from a tyrosine sidechain (TyrZ oxidation) and—in three of the four S-state transitions—manganese oxidation, until four electron holes (oxidizing equivalents) are accumulated by the Mn4Ca-oxo cluster in its S4 state. b, Example of tracing S-state transitions using IR absorption changes after excitation with visible-wavelength laser flashes (at zero on the time axis). The absorption changes (ΔA) are provided in optical density (OD) units. The IR transients at 1,384 cm−1 reflect symmetric stretching vibrations of carboxylate protein sidechains that sense changes in the oxidation state of manganese in the microsecond and millisecond time domain (coloured lines are simulations with time constants provided in Supplementary Table 2). Note that the scale on the x axis is linear below t = 0 and logarithmic above t = 0. c, The Mn4Ca cluster (Mn, violet; Ca, pink) in the S3 state with six bridging oxygens, the redox-active tyrosine (TyrZ), and further selected protein sidechains as well as water molecules (red spheres), based on crystal structures25. Assignment to polypeptide chains, numbering of the atoms of Mn4Ca-oxo and water molecules and hydrogen-bond distances are indicated in Supplementary Fig. 1. The two oxygens atoms that form the O–O bond in the oxygen-evolving S3 → S0 transition are indicated by red arrows.
Time-resolved tracking of O2 transition
To perform time-resolved infrared spectroscopy on PSII, we developed an FTIR step-scan experiment with automated exchange of dark-adapted PSII particles (Methods), thereby expanding previous experiments at individual wavenumbers towards detection of complete fingerprint spectra. The sample exchange system was refilled about every 60 h using PSII membrane particles with about 1.5 g of chlorophyll prepared from 40 kg of fresh spinach leaves for day and night data collection over a period of 7 months. We initiated the transitions between semi-stable S states by 10 visible light (532 nm) nanosecond laser flashes applied to the dark-adapted photosystems Using a specific deconvolution approach based on Kok’s standard model1 (Fig. 1a), we obtained time-dependent S-state difference spectra for each of the individual transitions between the four semi-stable reaction-cycle intermediates S1, S2, S3 and S0 (for selected time courses see Extended Data Fig. 2).
We focus on the oxygen-evolution transition, S3 → S4 → S0 + O2, predominantly induced by the third laser flash, for which time courses at selected wavenumbers are shown in F (time-resolved spectra are shown in Extended Data Fig. 3). Multiexponential simulations of the time courses provided 5 time constants describing acceptor- and donor-side PSII processes, including the expected time constants of 340 µs and 2.5 ms. The 2.5-ms time constant () corresponds to the reciprocal rate constant of the rate-determining step in O–O bond formation and O2 release8,9. The 340 µs time constant () corresponds to an obligatory step of proton removal from the oxygen-evolving complex of PSII, as shown recently by time-resolved detection of X-ray absorption, UV-visible spectroscopy, recombination fluorescence and photothermal signals resulting in a specific Mn(IV)4 metalloradical intermediate that was also trapped in low-temperature magnetic resonance experiments ‘Obligatory’ here signifies that the O–O bond formation chemistry can proceed only after proton removal is complete, as verified by the delayed onset of signals that trace manganese oxidation states or, generally, the O2 formation chemistry , which is also visible in the top time course of Fig. For systematic analysis of the 2D time–wavenumber data array obtained by the FTIR step-scan experiment, we exploited that the requirement for wavenumber independence of the time constants of proton removal ( = 340 µs) and the electron transfer associated with O2 formation ( = 2.5 ms), because they always reflect the same reaction (the same rate constant). The time constants can thus serve as a kinetic tag of the reaction in the time-resolved spectroscopic data. By simultaneous simulation of the time courses at 2,582 wavenumbers (1,800 cm−1 to 1,200 cm1) using the same set of time constants at each wavenumber, we obtained the amplitude spectra shown in Fig. , which are denoted as decay-associated spectra (DAS).

International Conference on Organic Chemistry https://organic-chemistry-conferences.sciencefather.com/
#environmentalchemistry #organicfarming #awards #chemistry #conference #inorganicchemistry #absorbent #biological #column Chromatography
Social Media Link :
Instagram: https://www.instagram.com/na.vy7928/
Twitter: https://twitter.com/AdinaAd27556369
Blogger: https://chemistry5225.blogspot.com/
Linkedin: https://www.linkedin.com/in/adina-adina-130a0b263/




No comments:
Post a Comment